(图文|赵虎 审核人|张红雨)6月29日,我校作物遗传改良国家重点实验室水稻团队谢为博教授课题组在Molecular Plant在线发表了题为“An inferred functional impact map of genetic variants in rice”的研究成果。该研究在准确获取4,726份水稻品种的单倍型图谱的基础上,定量注释了数百万个序列变异在水稻编码区以及调控区的效应,并详细分析了变异位点效应的功能特性及组织特异性,发现编码区大效应变异受到强烈的净化选择,而调控区大效应变异可能受到了正选择。该研究产生的序列变异功能效应图谱可用于筛选关键变异位点,将有助于水稻农艺性状重要基因的克隆和稀有优异变异的挖掘。
在过去的十年中,全基因组关联分析(GWAS)已被广泛用于水稻重要农艺性状关键调控基因的鉴定。然而,由于水稻中的连锁不平衡衰减距离较长,导致大量中性变异位点在GWAS中也很显著,因此关键变异位点的鉴定比较困难。同时,水稻中60%以上的变异位点的次要等位基因频率小于5%,这些稀有变异难以通过GWAS进行评估和挖掘。因此,构建一个准确、全面的功能注释平台来评估和预测序列变异的影响十分必要。这样的平台提供了独立于遗传学研究的信息,将可以帮助筛选GWAS关键变异位点,以及挖掘稀有优异变异位点。
该研究首先利用4,726份水稻品种的重测序数据鉴定出17,397,026个在至少10个品种中能重复鉴定到的变异位点,通过基因型填补,获得了各个品种准确完整的单倍型图谱。然后,基于编码区变异的单倍型信息鉴定了各品种的错义突变位点,并根据氨基酸残基的保存情况定量评估了每个错义突变的效应,最终得到了918,848个非冗余错义突变的效应。通过比较群体中具有不同等位基因频率的变异中大效应变异的比例,发现编码区的大效应突变倾向于具有较低的等位基因频率,即受到了强烈的净化选择。

图1 水稻序列变异效应图谱的构建框架
水稻中超过80%的变异位点位于非编码区,很多非编码区变异通过影响重要基因的表观状态和表达量进而影响了水稻的农艺性状。然而,这些变异的效应在之前的研究中一直缺乏关注和相关的研究手段。人类医学及该课题组最近的研究表明基于高质量的染色质可及性数据可建立深度学习模型,进而可以准确预测序列变异对染色质可及性的影响,据此可注释非编码区调控变异的效应(参见水稻团队发布基于深度学习模型预测植物基因组序列变异调控效应的网络服务)。
为此,该研究利用ATAC-seq技术首先获得了水稻品种珍汕97的6个代表性组织的高质量染色质可及性数据,从中鉴定出157,837个高可信的染色质开放区(open chromatin region,OCR),约占整个基因组的14.2%。发现很多重要基因的调控区及GWAS显著位点均富集于OCR中,表明OCR对于GWAS调控变异的鉴定十分重要。利用染色质可及性数据以及对应的基因组序列信息构建了深度神经网络模型。通过评估,该模型可以准确预测不同组织间的染色质开放状态,不同样本在测试集中的预测值与真实值相关性(R2)在0.61到0.72之间,各组织平均的曲线下面积(AUROC)达到0.945(图2)。利用杂种中等位基因特异的染色质可及性独立实验数据进行的评估表明,该模型预测单倍型效应方向的准确率达到0.94以上。这些结果表明该模型具有较高的准确性。
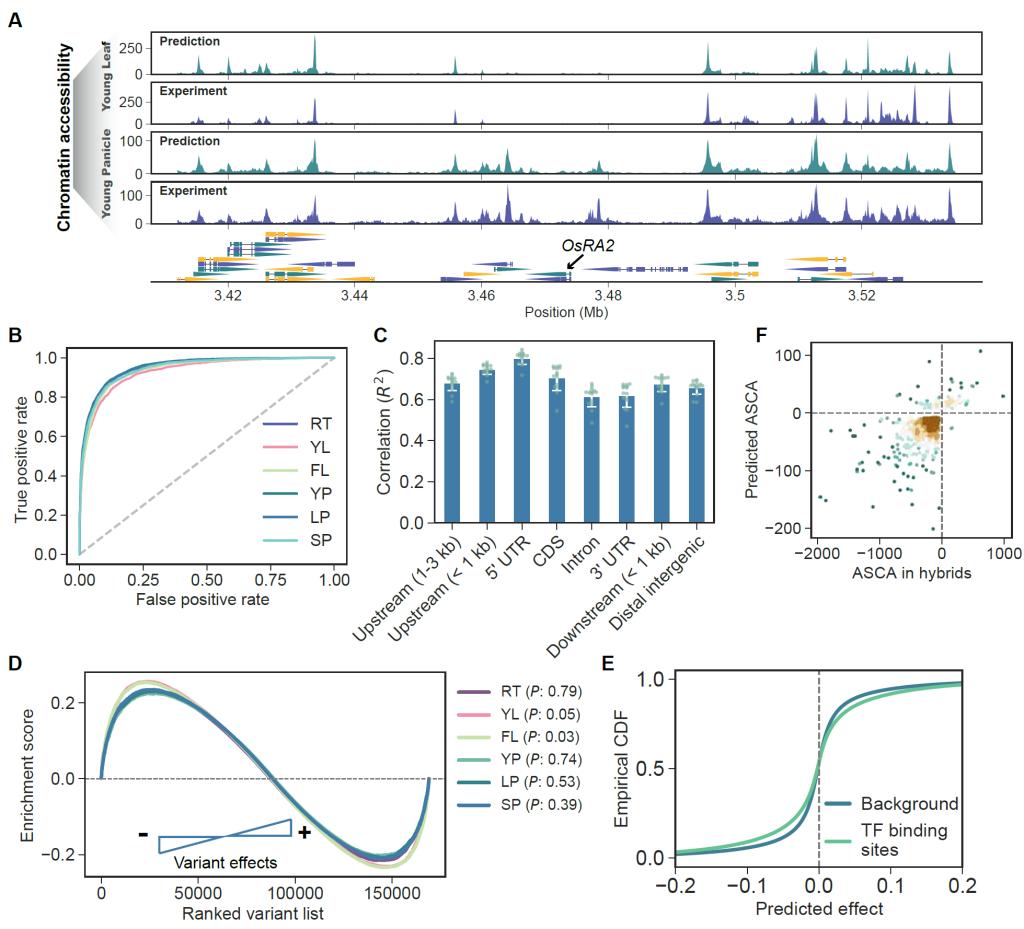
图2 深度学习模型准确性的评估
基于上述深度学习模型,该研究预测了5,067,405个序列变异对其周围染色质可及性的影响(效应)。与编码区大效应变异的分布不同的是,该研究发现水稻品种群体尤其是籼稻亚群在OCR区固定了大量的大效应调控变异,表明这些变异可能受到了正选择。通过与基因组分布比较发现这些大效应的变异在基因转录起始区域上游0-200 bp范围内拥有更高的比例。通过不同组织间变异效应的比较分析发现,变异效应在不同组织间有较高的一致性(R2在0.35到0.85之间),同时还发现组织间预测方向相反的大效应变异(在一个组织预测为突变后使得染色质可及性增加而在另一个组织中预测会使染色质可及性降低或相反)富集于基因间远端调控区域,而转录起始位点附近的启动子区域富集各个组织中方向相同的大效应变异。
该研究最后利用DEP1以及GW7等例子证明了该资源在寻找关键序列变异方面的可用性。为了方便更多研究者使用,该资源已被整合到RiceVarMap数据库中,使用者可以通过基因ID/名称以及变异ID、上传VCF文件等多种方式快速便捷的获取数据库中变异的注释结果。
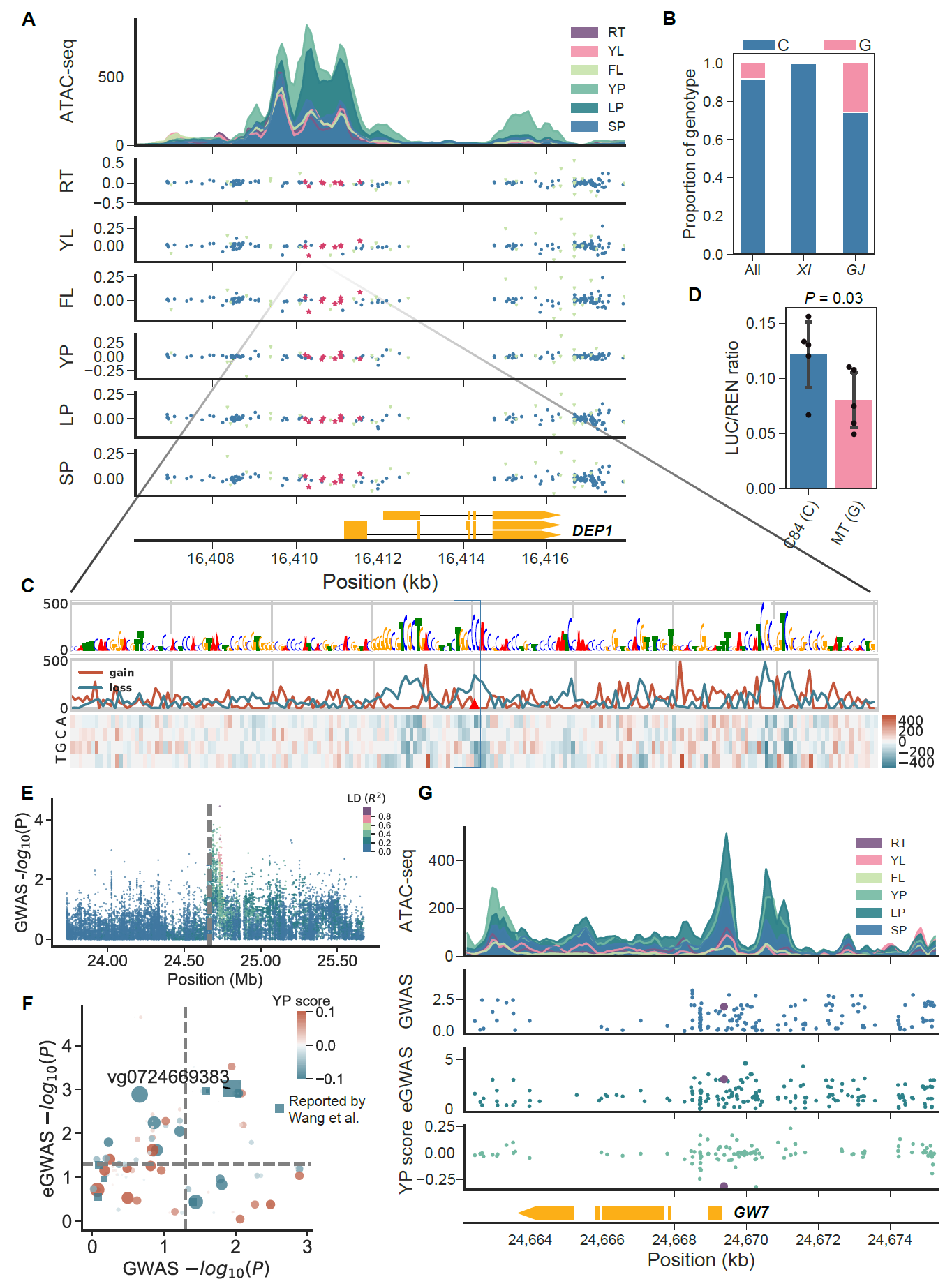
图3 序列变异效应图谱的使用示例
华中农业大学作物遗传改良国家重点实验室谢为博教授为该论文的通讯作者,赵虎博士为第一作者。广西大学陈玲玲教授以及华中农业大学熊立仲教授对该研究给予了指导和帮助。华中农业大学王磊博士以及倪福川老师也为本研究提供了帮助。该研究的数据分析工作得到武汉大学超算平台以及华中农业大学作物遗传改良国家重点实验室生物信息计算平台的支持和帮助,得到国家重点研发计划(2016YFD0100803)、国家自然科学基金(31821005, 31922065, 317717555)以及亚热带农业生物资源保护与利用国家实验室开放课题(SKLCUSA-b202002)的资助。
英文摘要
Interpreting the functional impacts of genetic variants (GVs) is an important challenge for functional genomic studies in crops and next-generation breeding. Currently, studies in rice (Oryza sativa) have mainly focused on the identification of GVs, while the functional annotation of GVs has not yet been systematically carried out. Here we present a functional impact map of GVs in rice. We curated haplotype information of 17,397,026 GVs from sequencing data of 4,726 rice accessions. We quantitatively evaluated the effects of missense mutations in coding regions in each haplotype based on the conservation of amino acid residues and obtained the effects of 918,848 non-redundant missense GVs. We also generated high-quality chromatin accessibility (CA) data from six representative rice tissues and used these data to train deep convolutional neural network models to predict the impacts of 5,067,405 GVs for CA in regulatory regions. We characterized the functional properties and tissue specificity of the effects of GVs and found that large-effect GVs in coding and regulatory regions might be subject to selection in different directions. We finally demonstrated how the functional impact map could be used to prioritize the causal variants in mapping populations. This impact map will be a useful resource for accelerating gene cloning and functional studies in rice and can be freely queried in RiceVarMap V2.0 .